

实验内容


实验环境
语言:Python
IDE:Pycharm
实验思路
步骤一
这一步其实就是筛选节点,得到图的点集,并把每个点都连边形成一个无向完全图。
每组数据行为基因,列为细胞。每个基因的表达方差其实就是这个基因的这一行的表达值的方差,然后从所有基因中筛选出方差最大的前 1000 个来作为图的节点集合,节点标识符定义为 ”基因名/CancerCell“ 或 ”基因名/TCell“。
每种癌症有两组数据,所以每种癌症会有两个无向完全图。
步骤二
这一步其实就是把上一步得到的六个无向完全图之间连边。
根据提供的 LigandReceptor_Human.txt 提供的受体配体来连边。
最终每种癌症的两个无向完全图会连在一起,形成一个新的无向完全图。
步骤三
这一步是为步骤一连的边加上边权。
根据要求计算边权即可。
spearman 相关系数可以直接用 pandas 库里的方法,效率要比 spicy 库的 spearmanr 函数快很多。
步骤四
这一步是为步骤二连的边加上边权。
根据要求计算边权即可。
步骤五
把每种癌症得到的无向图求最小生成树即可。
由于是稠密图,所以考虑使用 Prim 算法。
直接用库里的 get_mininum_spanning_tree 函数即可,效率要比自己手写的 MST 要高。
步骤六
把步骤五中得到的最小生成树中的 Intercellular Edges 提取出来,并把其两端延伸的五阶邻居也提取出来。
可使用库里的 single_source_shortest_path 函数来处理,比自己手写的 BFS 效率要高。
步骤七
对于每个最小生成树,求出其所有节点的度中心性,筛选出前 50 名。每个最小生成树的前 50 名比较异同,用韦恩图展示结果。
步骤八
对于每个最小生成树,求出其所有节点的介数中心性,筛选出前 50 名。每个最小生成树的前 50 名比较异同,用韦恩图展示结果。
代码
根据上述思路可得以下代码。
import os
import networkx as nx
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
from matplotlib_venn import venn3
from sklearn.preprocessing import minmax_scale
os.makedirs("Results", exist_ok=True)
def get_data(cancer, cell, base_path="InputData"):
file_path = os.path.join(base_path, f"{cancer}_{cell}_GeneExpression.csv")
df = pd.read_csv(file_path, index_col=0)
return df
def select_data(df):
var = df.var(axis=1)
selected = var.sort_values(ascending=False).head(1000).index
return df.loc[selected]
def build_intracellular_graph(df, cell):
graph = nx.Graph()
genes = df.index.tolist()
for gene in genes:
graph.add_node(f"{gene}/{cell}")
corr_matrix = df.T.corr(method='spearman').abs()
corr_matrix = corr_matrix.fillna(0)
upper_triangle = np.triu_indices_from(corr_matrix, k=1)
edges = [
(f"{genes[i]}/{cell}", f"{genes[j]}/{cell}")
for i, j in zip(upper_triangle[0], upper_triangle[1])
]
weights = 1 - minmax_scale(corr_matrix.values[upper_triangle])
for (u, v), w in zip(edges, weights):
graph.add_edge(u, v, weight=w)
return graph
def add_intercellular_edges(graph, cancer_df, tcell_df, ligand_receptor_file="LigandReceptor_Human.txt"):
ligand_receptor_df = pd.read_csv(ligand_receptor_file, sep='\t', header=None, names=['Ligand', 'Receptor'])
edges = []
res = []
for _, row in ligand_receptor_df.iterrows():
ligand, receptor = row['Ligand'], row['Receptor']
if ligand in cancer_df.index and receptor in tcell_df.index:
mean_ligand = cancer_df.loc[ligand].mean()
mean_receptor = tcell_df.loc[receptor].mean()
edges.append((f"{ligand}/CancerCell", f"{receptor}/TCell"))
res.append(mean_ligand * mean_receptor)
if ligand in tcell_df.index and receptor in cancer_df.index:
mean_ligand = tcell_df.loc[ligand].mean()
mean_receptor = cancer_df.loc[receptor].mean()
edges.append((f"{ligand}/TCell", f"{receptor}/CancerCell"))
res.append(mean_ligand * mean_receptor)
if res:
weights = 1 - minmax_scale(res)
for (u, v), w in zip(edges, weights):
graph.add_edge(u, v, weight=w)
def get_minimum_spanning_tree(graph):
return nx.minimum_spanning_tree(graph, weight='weight', algorithm='prim')
def get_intercellular_edges_and_neighbors(mst):
intercellular_edges = [(u, v) for u, v in mst.edges if u.split('/')[-1] != v.split('/')[-1]]
nodes = set()
for u, v in intercellular_edges:
nodes.update(nx.single_source_shortest_path(mst, u, cutoff=5).keys())
nodes.update(nx.single_source_shortest_path(mst, v, cutoff=5).keys())
return mst.subgraph(nodes)
def visualize_signal_pathway(graph, cancer):
plt.figure(figsize=(40, 24))
pos = nx.circular_layout(graph)
node_colors = []
for node in graph.nodes:
if node.endswith("/CancerCell"):
node_colors.append("red")
else:
node_colors.append("blue")
inter_edges = [(u, v) for u, v in graph.edges if u.split('/')[-1] != v.split('/')[-1]]
intra_edges = [(u, v) for u, v in graph.edges if u.split('/')[-1] == v.split('/')[-1]]
nx.draw_networkx_nodes(graph, pos, node_color=node_colors, node_size=30, alpha=0.8)
nx.draw_networkx_edges(
graph, pos,
edgelist=intra_edges,
edge_color="black",
width=0.5,
alpha=0.5
)
nx.draw_networkx_edges(
graph, pos,
edgelist=inter_edges,
edge_color="limegreen",
width=1.2,
alpha=0.8,
style="solid",
)
nx.draw_networkx_labels(
graph, pos,
font_size=6,
font_family='Arial',
font_weight='normal',
alpha=0.9
)
plt.title(f"{cancer} signal-pathway")
plt.axis('off')
plt.tight_layout()
plt.savefig(os.path.join("Results", f"{cancer}_signal_pathway.png"), dpi=300)
plt.close()
print(f"The signal pathway of {cancer} is done.")
def calculate_top_centrality_genes(graphs, centrality_type, top_n=50):
top_genes_list = []
for graph in graphs:
if centrality_type == "degree":
centrality = nx.degree_centrality(graph)
elif centrality_type == "betweenness":
centrality = nx.betweenness_centrality(graph, weight='weight')
sorted_genes = sorted(centrality.items(), key=lambda item: item[1], reverse=True)[:top_n]
top_genes = {gene for gene, score in sorted_genes}
top_genes_list.append(top_genes)
return top_genes_list
def plot_venn_diagram(sets, labels, title, filename):
plt.figure(figsize=(6, 6))
venn3(sets, set_labels=labels)
plt.title(title)
plt.tight_layout()
plt.savefig(os.path.join("Results", f"{filename}.png"), dpi=300)
plt.close()
def analyze_and_visualize_centrality(cancer_types, mst_list):
top_degree_genes = calculate_top_centrality_genes(mst_list, "degree")
plot_venn_diagram(
sets=top_degree_genes,
labels=cancer_types,
title="Degree Centrality Top 50 Genes Comparision",
filename="degree_centrality_venn"
)
print("The degree centrality venn is done.")
top_betweenness_genes = calculate_top_centrality_genes(mst_list, "betweenness")
plot_venn_diagram(
sets=top_betweenness_genes,
labels=cancer_types,
title="Betweenness Centrality Top 50 Genes Comparision",
filename="betweenness_centrality_venn"
)
print("The betweenness centrality venn is done.")
def main():
cancer_types = ["BreastTumor", "ColonTumor", "LungTumor"]
mst_list = []
for cancer in cancer_types:
print(f"Processing {cancer}...")
cancer_df = select_data(get_data(cancer, "CancerCell"))
tcell_df = select_data(get_data(cancer, "TCell"))
cancer_graph = build_intracellular_graph(cancer_df, "CancerCell")
tcell_graph = build_intracellular_graph(tcell_df, "TCell")
graph = nx.compose(cancer_graph, tcell_graph)
add_intercellular_edges(graph, cancer_df, tcell_df)
mst = get_minimum_spanning_tree(graph)
mst_list.append(mst)
subgraph = get_intercellular_edges_and_neighbors(mst)
visualize_signal_pathway(subgraph, cancer)
analyze_and_visualize_centrality(cancer_types, mst_list)
if __name__ == "__main__":
main()生成结果
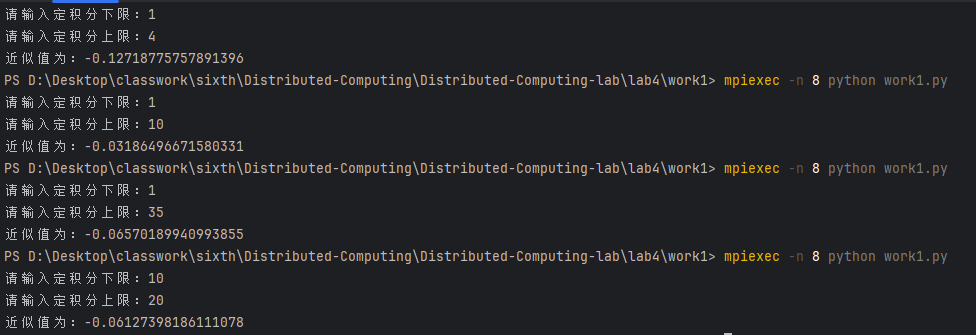
生成的图像结果如下。(图太大了直接传传不上来,所以压缩了一下,特别糊)








评论 (0)